发明简述
在本发明中,出人意料地发现,当将诸如酰氨基、脲基或氨基甲酸酯基的官能团引入到伯胺上时,可在一个步骤中以高产率制备7元环。此外,酰基能很容易除去,使得能在4位上进行精细设计。
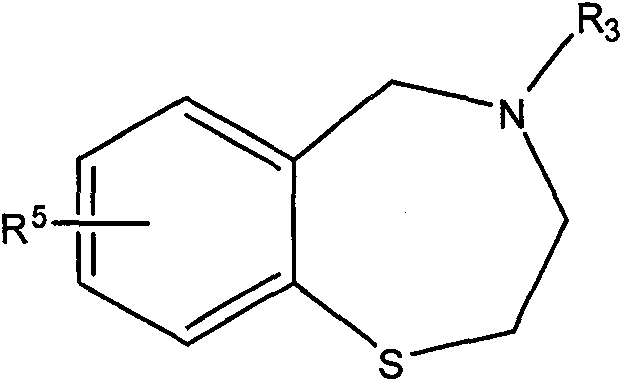
本发明涉及一种制备下式所示的2,3,4,5-四氢苯并[f][1,4]硫氮杂
的方法:
一方面,所述方法包括将下式所示的[2-(酰氨基乙基)硫基]芳烃:
与式R4CHO所示的醛或其多聚体以及酸反应。在这些结构式中,
Ar是单环、双环或三环的芳环或杂芳环体系;
R1、R2和R4各自独立地为H、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;
R3为C1-C10酰基、P(O)R8R9、C(=O)-R10、C(=S)-R11、S(=O)2R12、(CH2)mR13、氮保护基团、OH、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;或者R2和R3一起形成氧代取代的含氮杂环;
R5在其每次出现时独立地为H、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基、卤素、酰基、-SO3、-OR6、-SR6、-NR6aR6b、-N(R6)C(=O)OR7、N(R6)C(=O)R7、-C(=O)NR6aR6b、-C(=O)OR6、-C(=O)R6、-OC(=O)R6、-NO2、-CN、-C1-C6卤代烷基、-O-C1-C6卤代烷基、-N3或-P(O)R8R9,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;
R10和R11各自独立地为H、-OR14、-NR6aR6b、NHNHR15、NHOH、CONH2NHR15、CO2R15、CONR15、卤素、烷氧基、芳氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基、金刚烷氧基、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;
R6a、R6b、R12、R14、R15、R16和R17在其每次出现时独立地为H、-OR15、-NR15R16、NHNHR16、NHOH、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;或者R6a和R6b与它们所连接的氮一起表示5、6或7元含氮杂环;
R13是NH2、OH、-SO2R16、-NHSO2R16、C(=O)R17、NH(C=O)R17、-O(C=O)R17或-P(O)R8R9;m是1-10的整数;q是0或1-4的整数,条件是当R5为-C(=O)R6时,R5不处于硫侧链的邻位。
有利地,R3是乙酰基、苯甲酰基、甲苯甲酰基、苄氧基羰基、叔丁氧基羰基、丙烯酰基、乙二酰基或-C(=O)NR6aR6b,R5是C1-C4烃基、卤素、-OR6、-SR6、-NO2、-CN、-C1-C4卤代烷基或-O-C1-C4卤代烷基;R6是H或C1-C6烃基。另外,Ar优选为苯基。
或者,R
2和R
3一起形成吡咯烷酮、
唑烷酮或哌啶酮。在另一个实施方案中,R
1、R
2和R
4是氢。
在本发明的另一方面中,所述[2-(酰氨基乙基)硫基]芳烃可先与醛和碱反应,形成下式所示的[N-羟甲基-2-(酰氨基乙基)硫基]芳烃:
然后将所述[N-羟甲基-2-(酰氨基乙基)硫基]芳烃用酸处理,形成2,3,4,5-四氢苯并[f][1,4]硫氮杂
在本发明的两方面中,所述酸可为磺酸,如甲苯磺酸、苯磺酸、甲磺酸、对甲苯磺酸吡啶
盐或三氟甲磺酸,或路易斯酸,如醚合三氟化硼、四氯化钛、氯化铝或氯化锌;优选的醛或多聚体是甲醛、低聚甲醛或1,3,5-三
烷。在本发明的第二方面,所述碱可为一种或多种碱金属氢化物、氢氧化物或碳酸盐,吡啶或三烷基胺。碱的例子包括但不限于NaH、NaOH、KOH、Na
2CO
3、K
2CO
3、Cs
2CO
3、Et
3N或(iPr)
2NEt。
在本发明优选实施方案中,当R3是式-C(=O)-R18(其中R18是H、C1-C6烷基、C1-C6烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基)所示的C1-C10酰基、氮保护基团或OH时,下式所示的化合物:
被转化成下式所示的草酸化物:
其中M可为H、铵、碱金属或碱土金属。在所述特定的实施方案中,q是0或1;R1、R2和R4是氢;R3是式-C(=O)-R18所示的C1-C10酰基,其中R18是H、C1-C6烷基、C1-C6烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基;R5是H、C1-C4烃基、卤素、-OR6、-SR6、-NO2、-CN、-C1-C4卤代烷基或-O-C1-C4卤代烷基;R6是H或C1-C6烃基。
转化步骤典型地包括将R
3基团断开以提供下式所示的2,3,4,5-四氢苯并[f][1,4]硫氮杂
用草酸酯酰化所述2,3,4,5-四氢苯并[f][1,4]硫氮杂
然后将所述酯水解。在一个实施方案中,水解步骤包括用上文所用种类的碱对酯进行处理,并且任选地当M是H时,将其酸化。当M是H时,草酸酯化合物可进一步地转化成其盐,其中M是碱金属或碱土金属阳离子,如Na
+、Mg
++或Ca
++,或者M是铵,如NH
4 +。
在其中q是1且R
5是位于苯并硫氮杂
环7位的OCH
3的具体实施方案中,所述化合物如下式所示:
在该具体实施方案中,M可为氢、碱金属、碱土金属或铵。
在本发明的另一个实施方案中,下式所示的化合物
具体地以下式所示的[2-(酰氨基乙基)硫基]芳烃为起始物质而获得:
在该实施方案中,醛是低聚甲醛且酸是甲苯磺酸或盐酸。所述反应提供了下式所示的CBZ-保护的苯并硫氮杂
该方法进一步包括用酸将苄氧羰基断开以提供7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
用氯代草酸甲酯将所述7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
酰化;用碱的水溶液将所述甲酯水解;酸化以形成酸化合物;以及任选地将所述酸化合物转化为下式所示的化合物:
在该实施方案中,M是铵、碱金属或碱土金属。
在本发明的另一个实施方案中,2,3,4,5-四氢苯并[1,4]硫氮杂
通过如下方法而制备的:用式R
4CHO的醛或其多聚体以及酸对下式所示的[2-(酰氨基乙基)硫基]芳烃进行处理,
以制备下式所示的化合物:
在该实施方案中,R3是R3a,其中R3a是式-C(=O)-R18所示的C1-C10酰基,其中R18是H、C1-C6烷基、C1-C6烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基。
将所述R
3a取代基进一步断开以产生下式所示的4-未被取代的-2,3,4,5-四氢苯并[f][1,4]硫氮杂
用R
3X将所述4-未被取代的-2,3,4,5-四氢苯并[f][1,4]硫氮杂
酰化以制备下式所示的化合物:
在该实施方案中,R3是C1-C10酰基、P(O)R8R9、C(=O)-R10、C(=S)-R11、S(=O)2R12、(CH2)mR13、氮保护基团、OH、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;或者R2和R3一起形成氧代取代的含氮杂环;X是卤素、C1-C10酰氧基或活性酯基。在具体的实施方案中,R3是叔丁氧基羰基、苄氧基羰基、取代的苄氧基羰基或芴甲氧基羰基。在另一个实施方案中,R3X是酰基氯、酸酐、活性酯、氯甲酸酯或氨基甲酰氯。
在本发明的另一个实施方案中,将下式所示的化合物与式HN(R19)2的胺反应,
其中Ar、R1、R2、R4、R5和q定义如上,R3a是-C(=O)-R18,其中R18是C1-C4烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基;从而形成下式所示的化合物:
其中各R19为氢、烷基、芳基、杂环基、杂芳基、烷基芳基、烷基杂环基或烷基杂芳基,优选为H和C1-C6烷基,或N(R19)2一起表示5、6或7元含氮杂环。所述5、6或7元含氮杂环例如可为吡咯烷、哌啶、吗啉、4-CBZ哌嗪或氮杂环庚烷。
在本发明另一个优选的实施方案中,当[2-(酰氨基乙基)硫基]芳烃化合物具有下式所示的结构时:
它可与甲醛或其多聚体以及酸反应,形成下式所示的受保护的苯并硫氮杂
或者其可与甲醛或其多聚体以及碱反应,形成下式所示的[N-羟甲基-2-(酰氨基乙基)硫基]苯:
所述[N-羟甲基-2-(酰氨基乙基)硫基]苯可进一步与酸反应以提供下式所示的受保护的苯并硫氮杂
其中R3a是式-C(=O)-R18所示的C1-C10酰基;R5a是H、C1-C4烃基、卤素、-OR6、-SR6、-NO2、-CN、-C1-C4卤代烷基或-O-C1-C4卤代烷基;R6是H或C1-C6烃基;R18是H、C1-C6烷基、C1-C4烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基。
所述R3a基团可进一步断开以获得下式所示的化合物:
所述4-未被取代的-2,3,4,5-四氢苯并[f][1,4]硫氮杂
可用R
3X酰化以获得下式所示的化合物:
其中R3为C1-C10酰基;X是卤素、C1-C10酰氧基或活性酯基。
在另一个实施方案中,将下式所示的化合物
进一步转化为:
其中M是H、铵、碱金属或碱土金属。
在所述实施方案中,转化是通过将R
3a基团断开以提供2,3,4,5-四氢苯并[f][1,4]硫氮杂
用草酸酯酰化所述2,3,4,5-四氢苯并[f][1,4]硫氮杂
并水解所述酯而完成的。在一个实施方案中,所述水解步骤包括用碱处理所述酯,以及任选地当M是H时,将其酸化。任选地,将M是H的所得产物转化为其盐,在所述盐中M是阳离子如碱金属、碱土金属或铵。
在本发明的第三个方面中,[2-(酰氨基乙基)硫基]芳烃通过如下反应获得:将下式所示的化合物
与下式所示的化合物反应
从而得到下式所示的化合物:
然后与式R3X所示的化合物反应。可任选采用碱。LG是硫醇亲核取代的离去基团,X是胺亲核取代的离去基团。LG可为卤素,如氯、碘或溴,或磺酸酯,如甲磺酸酯、甲苯磺酸酯、苯磺酸酯、三氟甲磺酸酯、硝基苯磺酸酯或溴代苯磺酸酯。R3X典型地是酰基氯、酸酐、活性酯、氯甲酸酯或氨基甲酰氯。
在本发明的又一个方面中,[2-(酰氨基乙基)硫基]芳烃由下式所示的化合物
与下式所示的化合物反应制备
可任选采用碱。LG是硫醇亲核取代的离去基团,可为卤素(如氯、碘和溴),或磺酸酯(如甲磺酸酯、甲苯磺酸酯、苯磺酸酯、三氟甲磺酸酯、硝基苯磺酸酯和溴代苯磺酸酯)。
优选实施方案的详细描述
本发明涉及芳基稠合的四氢硫氮杂
,如2,3,4,5-四氢苯并[f][1,4]硫氮杂
的制备方法。
定义
在本说明书通篇中,术语和取代基的定义保持不变。
烷基旨在包括直链、支链或环状的烃基结构及其组合。低级烷基是指1至6个碳原子的烷基。低级烷基的例子包括甲基、乙基、丙基、异丙基、丁基、仲丁基和叔丁基等。优选的烷基是C20或更低的那些。环烷基是烷基的子集,包括3至8个碳原子的环状烃基。环烷基的例子包括环丙基、环丁基、环戊基和降莰基等。
C1至C20烃基包括烷基、环烷基、多环烷基、烯基、炔基、芳基及其组合。其例子包括苄基、苯乙基、环己基甲基、樟脑基和萘乙基。术语“碳环”旨在包括全部由任意氧化态的碳构成的环状体系。因此C3-C10碳环是指诸如环丙烷、苯和环己烯的体系;C8-C12碳多环是指诸如降冰片烷、萘烷、1,2-二氢化茚和萘的体系。
烷氧基是指通过氧与母结构相连的1至8个碳原子的直链、支链、环状构型及其组合的基团。其例子包括甲氧基、乙氧基、丙氧基、异丙氧基、环丙氧基和环己氧基等。低级烷氧基是指含有一至四个碳的基团。优选为甲氧基。对本申请的目的而言,烷氧基和低级烷氧基包括亚甲二氧基和亚乙二氧基。
氧代烷基是指其中一个或多个碳原子被氧所取代的烷基。其例子包括甲氧基丙氧基和3,6,9-三氧代癸基等。
酰基是指通过-C(=O)-相连并含有一至十个碳的取代基。所述基团还可含有杂原子如氧和氮。在一个实施方案中,酰基是指甲酰基以及通过一个羰基官能团与母结构相连并含有1至10个碳原子的直链、支链、环状构型的,饱和的、不饱和的和芳族及其组合的基团。酰基中的一个或多个碳原子可被氮、氧或硫取代,只要与母结构的连接点在羰基处即可。其例子包括乙酰基、苯甲酰基、丙酰基、异丁酰基、叔丁氧基羰基、苄氧基羰基和-C(=O)NH2等。低级酰基是指含有1至4个碳原子的基团。C1-C10酰基的例子也包括甲苯甲酰基、苄氧基羰基、叔丁氧基羰基、丙烯酰基、乙二酰基和-C(=O)N(R11)2,其中每个R11独立地为H或C1-C6烷基,或者N(R11)2一起表示5、6或7元含氮杂环。所述5、6或7元含氮杂环例如可为吡咯烷、哌啶、吗啉、4-CBZ哌嗪或氮杂环庚烷。本领域技术人员知晓,-C(=O)N(R11)2基团与其所连接的环氮原子(ring nitrogen)一起,也可命名为脲。在某些实施方案中,也可考虑T.W.Greene和P.G.M.Wuts在Protective Groups in Organic Synthesis[John Wiley&Sons,New York,1999]中描述的其他C1-C10酰基。
芳基(Ar)和杂芳基是指含有0-3个选自O、N或S的杂原子的5-或6-元芳环或杂芳环;含有0-3个选自O、N或S的杂原子的9-或10-元芳族双环体系或杂芳族环体系;或含有0-3个选自O、N或S的杂原子的13-或14-元芳族三环体系或杂芳族三环体系。芳族6-至14-元碳环包括例如苯、萘、1,2-二氢化茚、1,2,3,4-四氢化萘和芴;5-至10-元芳杂环包括例如咪唑、吡啶、吲哚、噻吩、苯并吡喃酮、噻唑、呋喃、苯并呋喃、苯并咪唑、喹啉、异喹啉、喹喔啉、嘧啶、吡嗪、四唑和吡唑。
芳烷基是指其中的芳基通过烷基与母结构相连的取代基。其例子为苄基和苯乙基等。杂芳烷基是指其中的杂芳基通过烷基与母结构相连的取代基。其例子包括例如吡啶基甲基和嘧啶基乙基等。
杂环是指其中1至3个碳原子被选自N、O和S的杂原子取代的环烷基或芳基。所述氮和硫杂原子可任选被氧化,氮杂原子可任选季铵化。落入本发明范围之内的杂环的例子包括吡咯烷、吡唑、吡咯、四氢异喹啉、苯并二
烷、苯并间二氧杂环戊烯(当作为取代基时,通常称作亚甲二氧基苯基)、四唑、吗啉、噻唑、吡啶、哒嗪、嘧啶、噻吩、呋喃、
唑、
唑啉、异
唑、二
烷和四氢呋喃等。值得注意的是,杂芳基是杂环的子集,其中杂环是芳基。
取代的烷基、芳基、环烷基和杂环基等是指各基团中至多三个H原子被卤素、卤代烷基、羟基、低级烷氧基、羧基、烷氧碳酰(也称作烷氧羰基)、酰胺基(也称作烷基氨基羰基)、氰基、羰基、硝基、氨基、烷基氨基、二烷基氨基、巯基、烷硫基、亚砜基、砜基、酰氨基、脒基、苯基、苄基、杂芳基、苯氧基、苄氧基或杂芳氧基取代的烷基、芳基、环烷基或杂环基。
术语“卤素”是指氟、氯、溴或碘。
在本发明通篇中,均出现了与“保护”、“脱保护”和“受保护的”官能团有关的术语。所述术语对本领域技术人员而言很容易理解,并用于涉及包括用一系列试剂连续进行处理的方法的段落中。在所述段落中,保护基团是指在工艺步骤中用于掩蔽某官能团的基团,在所述步骤中,所述官能团如不掩蔽将发生反应,而所述反应不是所期望的。保护基团能防止在所述步骤中发生反应,但是随后可能需要除去以将原官能团暴露出来。所述除去或“脱保护”在所述官能团可能造成干扰的一个或多个反应结束后进行。因此,如在本发明的方法中那样,当试剂的顺序确定后,本领域技术人员能够容易地想到适于作为“保护基团”的那些基团。
在本发明的情况下,必须保护的官能团包括胺,偶尔也包括羧酸和醇。用于该目的的适当的基团在化学领域的标准教科书中已有讨论,如T.W.Greene和P.G.M.Wuts的Protective Groups in Organic Synthesis[John Wiley&Sons,New York,1999],将其引入本文作为参考。特别需要注意的是标题为“Protection for the Amino Group”的这一章(494-614页)。
缩写Me、Et、Ph、Tf、Ts和Ms分别表示甲基、乙基、苯基、三氟甲磺酰基、甲苯磺酰基和甲磺酰基。有机化学家(即本领域技术人员)使用的缩写总列表可见于Journal of Organic Chemistry每一卷的第一期中。所述列单,典型地以标题为“Standard List of Abbreviations”的表的形式给出,本文将其引入本文作为参考。如本领域技术人员所理解的那样,术语“异丙醇”、“异丙基醇”和“2-丙醇”是等同的,均由CAS登记号67-63-0表示。
可用于本发明方法中的酸的例子包括但不限于磺酸或路易斯酸。磺酸的例子包括但不限于甲苯磺酸、苯磺酸、甲磺酸、三氟甲磺酸。路易斯酸的例子包括但不限于醚合三氟化硼、四氯化钛、氯化铝或氯化锌。酸性盐的例子是对苯甲磺酸吡啶
盐。
可用于本发明方法中的碱的例子包括但不限于碱金属氢化物、氢氧化物或碳酸盐,吡啶或三烷基胺。具体的碱包括NaH、NaOH、KOH、Na2CO3、K2CO3、Cs2CO3、Et3N和(iPr)2NEt。
在下文阐述的各实施方案中提到的酸或碱包括上文所提及的任意物质。
本发明的方法如方案1所示:
方案1
在本发明的第一个方面,本发明涉及一种制备下式所示的2,3,4,5-四氢苯并[f][1,4]硫氮杂
的方法:
所述方法形成了苯并硫氮杂环,并且包括用式R4CHO所示的醛或其多聚体,以及酸处理下式所示[2-(酰氨基乙基)硫基]芳烃的步骤:
在该方法中,Ar是单环、双环或三环芳环或杂芳环体系。其例子包括其中Ar是苯的化合物(苯并硫氮杂)以及其中Ar是例如萘、吡啶或苯并呋喃的化合物。
R1、R2和R4各自独立地为H、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代。
R3是C1-C10酰基、P(O)R8R9、C(=O)-R10、C(=S)-R11、S(=O)2R12、(CH2)mR13、氮保护基团、OH、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基、所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代。
R5在其每次出现时独立地为H、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基、卤素、酰基、-SO3、-OR6、-SR6、-NR6aR6b、-N(R6)C(=O)OR7、N(R6)C(=O)R7、-C(=O)NR6aR6b、-C(=O)OR6、-C(=O)R6、-OC(=O)R6、-NO2、-CN、-C1-C6卤代烷基、-O-C1-C6卤代烷基、-N3或-P(O)R8R9,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代。
R10和R11各自独立地为H、-OR14、-NR6aR6b、NHNHR15、NHOH、CONH2NHR15、CO2R15、CONR15、卤素、烷氧基、芳氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基、金刚烷氧基、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代。
R6a、R6b、R12、R14、R15、R16和R17在其每次出现时各自独立地为H、-OR15、-NR15R16、NHNHR16、NHOH、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代,或R6a和R6b与其所连接的氮一起表示5、6或7元含氮杂环。
R13是NH2、OH、-SO2R16、-NHSO2R16、C(=O)R17、NH(C=O)R17、-O(C=O)R17或-P(O)R8R9;m是1-10的整数;q是0或1-4的整数,条件是R5为-C(=O)R6时,R5不处于硫侧链的邻位。
这里的酰基是指通过-C(=O)-连接并含有一至十个碳的取代基。所述基团还可含有杂原子如氧和氮。C1-C10酰基的例子包括乙酰基、苯甲酰基、甲苯甲酰基、苄氧基羰基、叔丁氧基羰基、丙烯酰基、乙二酰基和-C(=O)N(R19)2,其中各个R19是氢、烷基、芳基、杂环基、杂芳基、烷基芳基、烷基杂环基或烷基杂芳基,或者N(R19)2一起表示5、6或7元含氮杂环。所述5、6或7元含氮杂环例如可为吡咯烷、哌啶、吗啉、4-CBZ哌嗪或氮杂环庚烷。在R3的子集中,R3a是式-C(=O)-R18所示的C1-C10酰基,其中R18可为H、C1-C4烷基、C1-C4烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基。在某些实施方案中,也可考虑T.W.Greene和P.G.M.Wuts在Protective Groups in Organic Synthesis中描述的其他C1-C10酰基。本领域技术人员知晓,-C(=O)N(R19)2基团与其相连的环氮原子一起,也可命名为脲。
作为另一种选择,R
2和R
3一起可形成氧代取代的含氮杂环。其例子包括吡咯烷酮、
唑烷酮或哌啶酮。在这些杂环中,氧与氮相邻,例如:
在醛的组分中,R
4可为H、C
1-C
6烷基、芳基、杂芳基、芳基C
1-C
6烷基和杂芳基C
1-C
6烷基。每个芳基和杂芳基都可任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代。当R
4是H时,所述醛是甲醛。甲醛本身为气体,因此作为其商购获得的低聚物和聚合物中的一种,即1,3,5-三
烷和低聚甲醛,通常更易操作。类似地,乙醛可采用其商购获得的三聚体,低聚醛(2,4,6-三甲基-1,3,5-三
烷),或者所述醛是低聚甲醛。本领域技术人员很清楚,可用任意醛的多聚体代替本发明方法中的醛。
碳环上的一个或多个取代基(R
5)
q可独立地为H、C
1-C
10烃基、卤素、-OR
6、-SR
6、-N(R
6)
2、-N(R
6)C(=O)OR
7、-C(=O)N(R
6)
2、-C(=O)OR
6、-C(=O)R
6、-OC(=O)R
6、-NO
2、-CN、-C
1-C
6卤代烷基、-O-C
1-C
6卤代烷基、-N
3或-P(O)R
8R
9。在这些取代基中,R
6可为H、C
1-C
10烃基、杂环基、杂环基-C
1-C
6烷基或芳基-C
1-C
6烷基;R
7可为C
1-C
10烃基、杂环基、杂环基烷基或芳基烷基;R
8和R
9可独立地为H、C
1-C
10烃基、杂环基、杂环基-C
1-C
6烷基或芳基-C
1-C
6烷基。在某些实施方案中,R
5可为H、C
1-C
4烃基、卤素、-OR
6、-SR
6、-NO
2、-CN、-C
1-C
4卤代烷基和-O-C
1-C
4卤代烷基;R
6可为H或C
1-C
6烃基。当R
5是-C(=O)R
6且处于硫侧链的邻位时,它可能干扰所期望的反应并降低苯并硫氮杂
的产率。
在本发明的第二个方面中,苯并硫氮杂
环在两步中而不是在一步中闭环。[2-(酰氨基乙基)硫基]芳烃与上述醛以及碱反应,从而形成下式所示的[N-羟甲基-2-(酰氨基乙基)硫基]芳烃:
将所述[N-羟甲基-2-(酰氨基乙基)硫基]芳烃用酸处理以形成2,3,4,5-四氢苯并[f][1,4]硫氮杂
在本发明的一个实施方案中,当R
3是式-C(=O)-R
18(其中R
18是H、C
1-C
6烷基、C
1-C
6烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基)所示的C
1-C
10酰基、氮保护基团或OH时,所述方法包括将下式所示的化合物:
转化成下式所示的化合物:
所述转化步骤通过将R
3基团断开以提供下式所示的2,3,4,5-四氢苯并[f][1,4]硫氮杂
完成:
用草酸酯将所述2,3,4,5-四氢苯并[f][1,4]硫氮杂
酰化;并除去所述酯。在一个实施方案中,通过水解除去所述酯。根据该实施方案,水解是通过用碱或酸处理所述酯而完成的。但是,在另一个实施方案中,当所述酯包含可通过加氢断开的官能团(如苄酯)时,苄基或其它可断开的基团可通过催化加氢除去,例如采用H
2和诸如Pd/C和Pt/C的金属催化剂。
M可为H、铵、碱金属(如钠)或碱土金属(如镁或钙)。术语“铵”旨在包括所有种类的氮阳离子化合物,包括精氨酸、NH4 +、N烷基H3 +、N(烷基)2H+、N(烷基)3H+和N(烷基)4 +。
本领域技术人员知晓,由本发明方法制备的化合物可为含水形式,如一水合物、脱水物和三水合物等,或者它们可为无水形式。类似地,由本发明方法制备的化合物可为与有机溶剂如醇形成的溶剂合物的形式,例如甲醇合物和乙醇合物等。
在一个实施方案中,所述方法如上文所述的那样采用其中R1、R2和R4是氢,R3是COOR的物质进行。将得到的酯断开以制备酸,所述酸可任选转化成其中M如上所述的盐。在其优选的实施方案中,M是钠。
在本发明的另一个实施方案中,所述方法包括制备下式所示的化合物:
其中[2-(酰氨基乙基)硫基]芳烃具有下式所示的结构:
所述醛是低聚甲醛,所述酸是甲苯磺酸或盐酸,并且所述反应提供了CBZ-保护的下式所示的苯并硫氮杂
用酸将所述苄氧羰基断开以提供7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
用氯代草酸甲酯将其酰化。用碱的水溶液将所述甲酯水解,然后将其酸化成酸化合物。
所述酸化合物任选被转化成下式所示的化合物:
其中M是铵,碱金属或碱土金属。
在本发明的另一个实施方案中,当其中R
3是R
3a时,2,3,4,5-四氢苯并[1,4]硫氮杂
通过用式R
4CHO所示的醛或其多聚体,以及酸处理下式所示的[2-(酰氨基乙基)硫基]芳烃制备:
从而制备下式所示的化合物:
其中R3a是式-C(=O)-R18所示的C1-C10酰基或氮保护基团,其中R18是H、C1-C6烷基、C1-C6烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基。
所述R
3a取代基可进一步断开以制备下式所示的4-未被取代的-2,3,4,5-四氢苯并[f][1,4]硫氮杂
进一步用R
3X将所述4-未被取代的2,3,4,5-四氢苯并[f][1,4]硫氮杂
酰化以制备下式所示的化合物:
取代基R3是C1-C10酰基、P(O)R8R9、C(=O)-R10、C(=S)-R11、S(=O)2R12、(CH2)mR13、氮保护基团、OH、(C1-C6)烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基、所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;并且X是卤素、C1-C10酰氧基或活性酯基。
在具体的实施方案中,所述R3取代基是叔丁氧基羰基、苄氧基羰基、取代的苄氧基羰基或芴甲氧基羰基;并且所述R3X是酰基氯、酸酐、活性酯、氯甲酸酯或氨基甲酰氯。
在具体的实施方案中,所述[2-(酰氨基乙基)硫基]芳烃化合物具有下式所示的结构:
并能与甲醛或其多聚体以及酸反应,从而形成下式所示的受保护的苯并硫氮杂
或与甲醛或其多聚体以及碱反应,从而形成下式所示的[N-羟甲基-2-(酰氨基乙基)硫基]苯:
所述[N-羟甲基-2-(酰氨基乙基)硫基]苯可进一步与酸反应以提供下式所示的受保护的苯并硫氮杂
所述取代基R3是式-C(=O)-R18所示的C1-C10酰基或氮保护基团,所述取代基R5a是H、C1-C4烃基、卤素、-OR6、-SR6、-NO2、-CN、-C1-C4卤代烷基或-O-C1-C4卤代烷基,所述取代基R6是H或(C1-C6)烃基;所述取代基R18是H、C1-C6烷基、C1-C4烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基。
进一步将其断开得到下式所示的化合物:
所述化合物可进一步用R3X酰化以得到下式所示的化合物
所述R3取代基是C1-C10酰基、P(O)R8R9、C(=O)-R10、C(=S)-R11、S(=O)2R12、(CH2)mR13、氮保护基团、OH、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基、所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;R10和R11各自独立地为H、-OR14、-NR6aR6b、NHNHR15、NHOH、CONH2NHR15、CO2R15、CONR15、卤素、烷氧基、芳氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基、金刚烷氧基、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代;R13是NH2、OH、-SO2R16、-NHSO2R16、C(=O)R17、NH(C=O)R17、-O(C=O)R17或-P(O)R8R9;m是1-10的整数;R6a、R6b、R12、R14、R15、R16和R17在其每次出现时各独立地为H、-OR15、-NR15R16、NHNHR16、NHOH、C1-C20烃基、C1-C6烷基、环烷基、芳基、杂环基、杂芳基、C1-C6烷基芳基、C1-C6烷基环烷基、C1-C6烷基杂环基或C1-C6烷基杂芳基,所述芳基、环烷基、杂环基和杂芳基每个都任选被一至三个独立地选自卤素、烷基、烷氧基、硝基、氰基和卤代烷基的取代基取代,或R6a和R6b与其所连接的氮一起表示5、6或7元含氮杂环;X是卤素、C1-C10酰氧基或活性酯基。
在另一个实施方案中,进一步将下式所示的化合物:
转化成下式所示的化合物:
其中M是H、铵、碱金属或碱土金属,所述转化通过将R
3a基团断开以提供2,3,4,5-四氢苯并[f][1,4]硫氮杂
用草酸酯将所述2,3,4,5-四氢苯并[f][1,4]硫氮杂
酰化,并水解所述酯而完成。
所述水解步骤包括用碱或酸处理所述酯,并且任选地,当M是H时,将其酸化。任选地将得到的M为H的产物被转化为其盐。M可为阳离子如Na+、Ca++、Mg++或铵。
用于一步和两步闭环反应中的[2-(酰氨基乙基)硫基]芳烃化合物可通过将下式所示的化合物:
与下式所示的化合物:
反应,以提供下式所示的化合物而获得:
该反应任选在碱的存在下进行。LG是硫醇亲核取代的离去基团。硫醇取代的离去基团是本领域已知的。它们包括卤素和磺酸酯。其例子包括氯、碘、溴、甲磺酸酯、甲苯磺酸酯、苯磺酸酯、三氟甲磺酸酯、硝基苯磺酸酯和溴代苯磺酸酯。
在另一个实施方案中,所述[2-(酰氨基乙基)硫基]芳烃可通过将下式所示的化合物:
与下式所示的化合物:
反应,以提供下式所示的化合物而获得:
该反应任选在碱的存在下进行。所述酰基取代基R3可通过将式R3X所示的化合物,任选在碱的存在下,在第二步反应中加成。LG是硫醇亲核取代的离去基团,X是胺亲核取代的离去基团。因此R3X可为酰基氯、酸酐、活性酯、氯甲酸酯、磺酰氯或氨基甲酰氯。“活性酯”是本领域,特别是肽合成领域所公知的术语,是指能够与胺发生取代反应以形成酰胺的酯。所述术语包括被相邻吸电子取代基“活化”的酯。其例子包括酚类的酯,特别是电负性取代基取代的苯酚酯如五氟苯酚酯;异脲的O-酯,如通过与碳化二亚胺相互作用而获得的那些;N-羟基酰亚胺和N-羟基杂环化合物的O-酯。其具体例子包含S-叔丁酯、S-苯酯、S-2-吡啶酯、N-羟基哌啶酯、N-羟基琥珀酰亚胺酯、N-羟基邻苯二甲酰亚胺酯和N-羟基苯并三唑酯。
在另一个实施方案中,本发明包括一种制备2,3,4,5-四氢苯并[1,4]硫氮杂的方法,所述方法包括将R3基团断开以制备下式所示的化合物:
然后任选在碱的存在下与式R3X所示的化合物反应;其中X是胺亲核取代的离去基团。所述R3X选自酰基氯、酸酐、活性酯、氯甲酸酯和氨基甲酰氯。
在一个特定的实施方案中,将下式所示的化合物:
与式X-CH2-COOR’所示的化合物反应,其中X是卤素,R’是烷基,从而形成下式所示的酯类化合物
然后通过将所述酯水解以形成下式所示的化合物:
在一个特定的实施方案中,本发明提供了制备下式所示化合物的方法:
其中Ar、R1、R2、R4、R5和q定义如上;R3是-C(=O)-R20,其中R20是N(R21)2,其中各R21独立地选自氢、烷基、芳基、杂环基、杂芳基、烷芳基、烷基杂环基或烷基杂芳基,或N(R21)2一起表示5、6或7元含氮杂环。所述5、6或7元含氮杂环例如可为吡咯烷、哌啶、吗啉、4-CBZ哌嗪或氮杂环庚烷。根据该实施方案,所述方法包括将下式所示的化合物:
与式HN(R19)2所示的胺反应,其中Ar、R1、R2、R4、R5和q定义如上,R3是-C(=O)-R21,其中R21是C1-C4烷氧基、烯丙氧基、苄氧基、取代的苄氧基、芴甲氧基或金刚烷氧基,所述胺中的各R19独立地如上述定义,从而形成下式所示的化合物:
采用本发明的方法,该实施方案具有如下优点:简单并且能获得高氨基甲酸酯合成产率,并且在简单、高效且能以高产率获得高品质的产物的工艺中,将所述氨基甲酸酯进一步转化成脲衍生物。
对本领域的技术人员而言,很明显下式所示的化合物:
可用作制备各种1,4-苯并硫氮杂化合物的起始物质-,如美国专利申请11/506,285(US 2007/0173482)、11/212,413(US2007/0049572)、11/212,309(US2006/0194767)和10/809,089(US2005/0215540)中描述的那些,将其全部内容引入本发明以作为参考。
实施例
下文将对落入本发明范围之内的示例性方法加以描述。
实施例1:2-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)-2-氧代乙酸-(6)的合成(方案2)。
方案2
2-(4-甲氧基苯硫基)乙胺(1)
将4-甲氧基苯硫酚(50g,0.357mol),2-氯乙胺单盐酸盐(39.8g,0.343mol),K2CO3(78.8g,0.57mol)和二异丙基乙胺(32mL,0.178mol)在200mL四氢呋喃中混合。将混合物减压脱气5分钟,在氩气下回流过夜。除去溶剂并向烧瓶中加入水(300mL)。将混合物用二氯甲烷萃取(3×200mL)。收集有机相,除去二氯甲烷,加入50mL浓盐酸,然后加入200mL水。将该溶液用1∶1的EtOAc/己烷萃取(3×200mL)。用2M的NaOH将水层的pH值调节至10,用二氯甲烷萃取(3×200mL)。合并的有机溶液用无水硫酸钠干燥。除去溶剂得到61g无色液体状的目标化合物,产率为97%。
1H-NMR(300MHz,CDCl3):7.35(d,J=8.7Hz,2H),6.81(d,J=8.7Hz,2H),3.77(s,3H),2.88-2.80(m,4H),1.44(s,2H)。
2-(4-甲氧基苯硫基)乙基氨基甲酸苄酯(2)
方法一:
在0℃下,向装有化合物1(8.0g,43.7mmol)、碳酸氢钠(12.1g,144mmol)、水(100mL)和二氯甲烷(200mL)的烧瓶中滴加氯甲酸苄酯(8.2g,48.1mmol,稀释在100mL二氯甲烷中)。加完后,将该混合物室温搅拌5小时。收集有机层,水溶液用100mL二氯甲烷萃取。合并的有机溶液用硫酸钠干燥。除去溶剂,得到的固体用200mL的四氢呋喃/己烷(1∶10)研制。收集固体并干燥,得到目标产物(12.9g),产率为93%。
另一种方法:
在0℃下,向化合物1(10g,54.6mmol)和三乙胺(15mL,106mmol)的200mL二氯甲烷溶液中滴加氯甲酸苄酯(7.24mL,51.5mmol,稀释在100mL二氯甲烷中)。加完后,将混合物在室温搅拌1小时。过滤除去固体。溶液用100mL的0.1M HCl和100mL饱和碳酸钠萃取,并用无水硫酸钠干燥。除去溶剂,得到的白色固体在200mL的四氢呋喃/己烷(1∶20)中搅拌3小时。过滤收集所述固体,得到14.2g目标化合物,产率为87%。
1H-NMR(300MHz,CDCl3):7.35(m,7H),6.83(d,J=8.7Hz,2H),5.07(m,3H),3.77(s,3H),3.10(q,J=6.3Hz,2H),2.92(t,J=6.3Hz,2H)。
7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(3)
将处于250mL甲苯中的化合物2(7.3g,23mmol)、低聚甲醛(6.9g,0.23mol)和对甲苯磺酸(1.45g,7.6mmol)的混合物在70℃下搅拌过夜。冷却至室温后,滤掉固体。将溶液用饱和碳酸钠(100mL)萃取,并用无水硫酸钠干燥有机层。除去溶剂后,获得液态目标产物(7.4g),产率为97%。
1H-NMR(300MHz,CDCl3):7.44(d,J=8.1Hz,0.77H),7.32(m,5.60H),7.07(d,J=2.7Hz,0.33H),6.68(m,1.30H),5.04(s,2H),4.59(ss,2H),3.96(br,1.80),3.80(ss,1.23H),3.55(s,1.97H),2.76(m,2H)。
7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
氢溴化物(4HBr盐)
方法一:
将HBr溶液(在乙酸中的浓度为33%,10mL)加入到化合物3(4.2g,12.8mmol)中。加完后,开始产生二氧化碳并形成白色固体。将该混合物再室温静置2小时。向所述混合物中加入乙醚(150mL),并搅拌30分钟。过滤收集固体并用乙醚洗涤。将所述固体在真空下干燥,获得3.40g目标化合物,产率为91.8%。
1H-NMR(300MHz,DMSO-d6):9.02(br,2H),7.52(d,J=8.1Hz,1H),7.27(d,J=3.3Hz,1H),6.92(dd,J=8.4,2.7Hz,1H),4.41(s,2H),3.77(s,3H),3.53(m,2H),2.96(m,2H)。
另一种方法(游离碱4a):
将化合物3(10g,30mmol)与50mL浓HCl、50mL水和30mL二噁烷混合。将该混合物在100℃下搅拌过夜。冷却至室温后,减压除去大部分溶剂和HCl。向所述溶液加入水(100mL),滤除固体。水溶液用EtOAc/己烷(1∶1,3×100mL)萃取,加入15g NaOH碱化。混合物用二氯甲烷(3×150mL)萃取。合并的溶液用无水硫酸钠干燥。除去溶剂后得到液体,将其室温静置后凝固,获得6.2g目标化合物。
1H-NMR(300MHz,CDCl3):7.42(d,J=8.1Hz,1H),6.78(d,J=2.7Hz,H),6.68(dd,J=2.7,8.1Hz,1H),4.08(s,2H),3.96(br,1.80),3.76(s,3H),3.38(m,2H),2.68(m,2H)。
2-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)-2-氧代乙酸甲酯(5)
向化合物4(580mg,2.97mmol)和二异丙基乙胺(1.0mL,5.5mmol)的20mL二氯甲烷溶液中加入氯代草酸甲酯(301μl,3.27mmol)。将该溶液在室温下搅拌4小时。用40mL二氯甲烷稀释并用1M HCl(2×30mL)萃取。将有机层用硫酸钠干燥。除去溶剂得到目标化合物(740mg),产率为89%。
1H-NMR(300MHz,CDCl3):7.46(d,J=8.4Hz,1H),7.09(d,J=2.7Hz,1H),6.73(m,1H),4.76(br,2H),4.06(m,0.6H),3.87(m,7.4H),2.81(m,2H)。
2-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)-2-氧代乙酸(6)
将化合物5(740mg)溶解到四氢呋喃、甲醇和1M的NaOH的30mL混合物(1∶1∶1)中。将该溶液在室温下搅拌6小时并用1N的盐酸酸化。除去有机溶剂,收集得到的固体并用水洗涤。将所述固体真空干燥,获得600mg固体,产率为85%。
1H-NMR(300MHz,DMSO-d6):7.43(m,2H),7.00(d,J=2.7Hz,1H),6.79(m,1H),4.66(ss,2H),3.98-3.82(m,2H),3.73(ss,3H),2.83(m,2H)。
实施例2:化合物38(例如,7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)(哌嗪-1-基)甲酮(ARM064))的合成(方案3)。
方案3
化合物1与ClC(=O)OR反应形成化合物36,其中R是苯基、4-NO2-苯基、甲基、乙基、苄基、烯丙基、CH2CCl3,CH2CF3或者在与胺反应时能与其所连接的氧一起作为离去基团的其他基团。化合物36在酸中与(CH2O)n反应形成化合物37。将化合物37和胺(HNR”)的混合物在碱或催化剂如Al(CH3)3(Janda,K.D.等,Tetrahedron 2004,60,3439)或γ-Al2O3(Vauthey,I.等,Tetrahedron Lett.2000,41,6347)的存在下加热,得到化合物38。或者化合物37与胺(HNR”)的反应可采用金属催化剂如Zr(Ot-Bu)4(Porco,J.A.等,Organic Lett.2007,1517)催化。为了制备ARM064,HNR”是哌嗪或Boc保护的哌嗪。当使用Boc保护的哌嗪时,Boc基团可在诸如TFA的酸中除去。得到的化合物38是ARM064,产率:85%-95%。
实施例3:化合物38(例如7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)(哌嗪-1-基)甲酮(ARM064))的合成(方案4)。
方案4
化合物1与ClC(=O)OR反应形成化合物36,其中R是苯基、4-NO2-苯基、甲基、乙基、苄基、烯丙基、CH2CCl3,CH2CF3或者在与胺反应时能作为离去基团的其它基团。化合物36在酸中与(CH2O)n反应,形成化合物37。然后将氨基甲酸酯基除去得到游离胺,即化合物4a。当R为苄基时,如实施例1中将化合物3转化为化合物4和4a所述的那样,能将氨基甲酸酯37转化成化合物4或其游离碱化合物4a。化合物4a(或4)与三光气反应,然后任选在碱的存在下与胺(HNR”)反应。为制备ARM064,HNR”优选为Boc保护的哌嗪。当使用Boc保护的哌嗪时,所述Boc基团可在诸如TFA酸中除去。得到的化合物38是ARM064。
实施例4:7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸甲酯(8)的合成(方案5)。
方案5
2-(4-甲氧基苯硫基)乙胺盐酸盐(1-HCl)
将处于DMF(200mL)中的4-甲氧基苯硫酚(25g)、2-氯乙胺盐酸盐(1.1当量)和Cs2CO3(2.4当量)的混合物在60℃下搅拌2天。在减压下蒸发除去溶剂,将粗产物溶于300mL的EtOAc中。有机相用水(2×50mL)洗涤并减压浓缩。将剩余物溶于200mL 1N的HCl中,用EtOAc(2×50ml)洗涤。蒸发水相得到所期望的产物的盐酸盐,其为白色固体。产率:34.8g,91%。(用1N的NaOH处理所述盐可获得游离胺)。
2-(4-甲氧基苯硫基)乙基氨基甲酸甲酯(7)
向冷却至约0℃的化合物1-HCl(0.466g)的CH2Cl2(20mL)溶液中加入氯甲酸甲酯(1.1当量)和三乙胺(2.5当量)。将该反应混合物在0℃下搅拌2小时,并用1N的HCl和饱和NaHCO3洗涤。在减压下蒸发除去溶剂,获得白色固体状的产物7,产率:480mg,98%。
1N-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),5.1(宽,1H),3.8(s,3H),3.65(s,3H),3.30(t,2H),2.95(t,2H)。
7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸甲酯(8)
将处于苯(4mL)中的化合物7(95mg)、低聚甲醛(95mg,过量)、对甲苯磺酸(30mg)的混合物在60℃下搅拌过夜。TLC分析显示出现了一个新斑点,并且起始物质完全消失了。过滤,然后浓缩并经SiO2层析得到92mg纯净的产物8,产率:95%。
1H-NMR(300MHz,CDCl3):δ:7.40(d,0.6H),δ:7.38(d,0.4H),6.98(s,0.4H),6.80(s,0.6H),6.61(d,1H),4.35(s,0.4×2H),4.30(s,0.6×2H),3.89(s,宽,2H),3.70(s,3H),3.60(s,0.6×3H),2.90(m,2H)。
实施例5:7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸甲酯(8)的合成(方案6)。
方案6
羟甲基(2-(4-甲氧基苯硫基)乙基)氨基甲酸甲酯(9)
将处于THF(50mL)中的化合物7(2.0g)、低聚甲醛(1.5g)、Cs2CO3(1.2当量)的混合物在70℃下搅拌3小时。过滤并浓缩溶剂,获得目标产物9的纯净白色固体,产率:2.2g,98%。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.89(d,2H),4.7(宽,2H),3.75(s,3H),3.65(s,3H),3.40(t,2H),2.95(t,2H)。
7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸甲酯(8)
在Ar下,用1.1当量的BF
3-OEt
2处理化合物9(20.0mg)的10mL CH
2Cl
2溶液。将该混合物搅拌过夜,用1N的HCl和饱和NaHCO
3洗涤。除去溶剂得到粗产物,TLC分析显示存在一个主要的斑点。将所述粗产物进一步经SiO
2柱提纯,获得目标化合物(16mg)。其结构通过与可信样品的NMR和TLC进行对比而确定,所述可信样品通过7-甲氧基-2,3,4,5-四氢-1,4-苯并硫氮杂
与氯甲酸甲酯的反应制备。
1H-NMR(CDCl3):δ:7.40(d,0.6H),δ:7.38(d,0.4H),6.98(s,0.4H),6.80(s,0.6H),6.61(d,1H),4.35(s,0.4×2H),4.30(s,0.6×2H),3.89(s,宽,2H),3.70(s,3H),3.60(s,0.6×3H),2.90(m,2H)。
实施例6:1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)乙酮(11)的
合成(方案7)
方案7
N-(2-(4-甲氧基苯硫基)乙基)乙酰胺(10)
在冷却至约0℃的化合物1-HCl(0.44g)的CH2Cl2(15mL)溶液中加入乙酰氯(1.0mL)和三乙胺(1.0mL)。将该反应混合物在0℃下搅拌1小时,并用1N的HCl和饱和NaHCO3洗涤。通过减压蒸发除去溶剂,获得产物10的固体(TLC和NMR表明其是纯的),产率0.5g。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),5.90(s,宽,1H),3.8(s,3H),3.40(t,2H),2.95(t,2H),2.1(s,3H)。
1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)乙酮(11)
将处于苯(4mL)中的化合物11(70mg)、低聚甲醛(70mg)、对甲苯磺酸(40mg)的混合物在70℃下搅拌过夜。过滤,然后用饱和NaHCO3溶液洗涤获得粗产物,所述粗产物经SiO2层析法提纯,获得目标化合物(11),产率为91%。
1H-NMR(300MHz,CDCl3):δ:7.50(d,0.6H),δ:7.38(d,0.4H),7.10(s,0.4H),6.80(s,0.6H),6.70(m,1H),4.70(s,0.4×2H),4.60(s,0.6×2H),4.10(s,宽,0.6×2H),3.90(s,宽,0.4×2H),3.80(s,0.4×3H),3.79(s,0.6×3H),2.90(m,2H),2.2(s,0.4×3H),2.06(s,0.6×3H)。
实施例7:2,2,2-三氟-1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)乙酮(13)的合成(方案8)
方案8
2,2,2-三氟-N-(2-(4-甲氧基苯硫基)乙基)乙酰胺(12)
将化合物1(盐酸盐,1.8g)的CH2Cl2(20mL)溶液冷却至约4℃,加入三氟乙酸酐(1.1当量)和三乙胺(1.5当量)。将该反应混合物在室温下搅拌过夜,用1N的HCl和饱和NaHCO3洗涤。通过减压蒸发除去溶剂获得固体产物12(TLC和NMR表明其是纯的),产率:2.5g。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),6.6(s,宽,1H),3.8(s,3H),3.40(t,2H),2.95(t,2H)。
2,2,2-三氟-1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)乙酮(13)
将处于甲苯(10mL)中的化合物12(100mg)、低聚甲醛(100mg)和对甲苯磺酸(60mg)的混合物在80℃下搅拌过夜。过滤,然后用饱和NaHCO3溶液洗涤得粗产物13,用TLC和NMR估算产率为约70%。
1H-NMR(300MHz,CDCl3):δ:7.45(d,1H),7.05(s,1H),6.7(d,1H),4.6(m,2H),4.0(m,2H),3.8(s,3H),2.9(m,2H)。
实施例8:7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲醛(15)的合成(方案9)
方案9
N-(2-(4-甲氧基苯硫基)乙基)甲酰胺(14)
将化合物1(550mg)和Et3N(1mL)的HCO2Et(20mL)溶液回流12小时。将该混合物冷却并用1N的HCl和饱和NaHCO3洗涤。通过减压蒸发除去溶剂,获得固体产物14:606mg。TLC和NMR表明所述粗产物是纯的,不需要进一步提纯就可用于下一步反应。
1H-NMR(300MHz,CDCl3):8.15(s,1H),7.4(d,2H),6.8(d,2H),6.2(s,宽,1H),3.8(s,3H),3.40(t,2H),2.95(t,2H)。
7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲醛(15)
将处于苯(5mL)中的化合物14(100mg)、低聚甲醛(100mg)、对甲苯磺酸(60mg)的混合物在70-75℃下搅拌2天。过滤,然后用饱和NaHCO3溶液洗涤,浓缩得粗产物15,用TLC和NMR估算产率为约70%。
1H-NMR(CDCl3):δ:8.2(s,1H),7.50(d,1H),7.10(s,1H),6.80(d,1H),4.80(s,宽,2H),4.10(s,宽,2H),3.80(s,3H),2.90(m,2H)。
实施例9:(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)(吗啉代)甲酮(17)的合成(方案10)
方案10
N-(2-(4-甲氧基苯硫基)乙基)吗啉-4-甲酰胺(16)
在0℃下,将三光气(1.0当量)和三乙胺(2.5当量)加入到化合物1(400mg)的CH2Cl2(10mL)溶液中。将该反应混合物在室温搅拌2天,加入吗啉(3.0当量)。在搅拌下反应直至TLC显示反应完全(约4小时),用1N的HCl和饱和NaHCO3(3×10mL)洗涤。通过减压蒸发除去溶剂,获得产物16(520mg)。TLC和NMR表明粗产物是纯的,不需要进一步提纯就可用于下一步反应。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),3.75(s,3H),3.65(m,4H),3.40(t,2H),3.28(m,4H),2.95(t,2H)。
(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)(吗啉代)甲酮(17)
将处于苯(5mL)中的化合物16(100mg)、低聚甲醛(110mg)、对甲苯磺酸(60mg)的混合物在70-75℃下搅拌14小时。将反应溶液过滤,用饱和NaHCO3溶液洗涤,浓缩得目标粗产物17,用TLC和NMR估算产率为约50%。经SiO2层析法(CH2Cl2/EtOAc 10∶1)提纯,获得了用于结构确认的纯试样。
1H-NMR(300MHz,CDCl3):δ:7.4(d,1H),6.95(s,1H),6.7(d,1H),4.5(s,2H),3.80(s,3H),3.79(s,宽,2H),3.70(m,4H),3.10(m,4H),2.95(t,2H)。
实施例10:4-(7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
-4-羰基)哌嗪-1-甲酸苄酯(19)的合成(方案11)
方案11
4-(2-(4-甲氧基苯硫基)乙基氨基甲酰基)哌嗪-1-甲酸苄酯(18)
在0℃下,将三光气(0.7当量)和三乙胺(1.0当量)加入到化合物1(183mg)的CH2Cl2(10mL)溶液中。将该反应混合物在室温搅拌1小时,然后加入N-CBZ-哌啶(1.5当量)和Et3N(0.5mL)。将反应混合物搅拌过夜,用1N的HCl和饱和NaHCO3(3×5mL)洗涤。通过减压蒸发除去溶剂,获得目标产物18,用层析法提纯(SiO2,CH2Cl2/EtOAc 10∶1)。产率:360mg。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),7.30(s,5H),6.8(d,2H),6.6(s,宽,1H),5.15(s,2H),3.75(s,3H),3.50(m,4H),3.40(t,2H),3.30(m,4H),2.95(t,2H)。
4-(7-甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
-4-羰基)哌嗪-1-甲酸苄酯(19)
将处于苯(5mL)中的化合物18(30mg)、低聚甲醛(100mg)和对甲苯磺酸(30mg)的混合物在70-75℃下搅拌2天。将反应混合物过滤,用饱和NaHCO3溶液洗涤,浓缩得粗产物19,用SiO2层析法提纯(CH2Cl2/EtOAc10∶1),产率:21mg,69%。
1H-NMR(300MHz,CDCl3):δ:7.4(d,1H),7.35(s,5H),6.85(s,1H),6.70(d,1H),5.15(s,2H),4.5(s,2H),3.80(s,3H),3.75(s,宽,2H),3.60(m,4H),3.20(m,4H),2.95(t,2H)。
实施例11:8-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(22)和6-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(23)的合成(方案12)
方案12
2-(3-甲氧基苯硫基)乙胺(20)
将处于在CH3CN(60mL)中的3-甲氧基苯硫酚(5g)、2-氯乙胺盐酸盐(1.1当量)和Cs2CO3(2.2当量)的混合物在60℃下搅拌20小时。向反应混合物中加入EtOAc(100mL),用水(2×30mL)洗涤。减压浓缩有机相,获得产物20(产率:6.2g,95%),TLC和NMR表明所述产物是纯的,不需要进一步提纯就可用于下一步反应。
2-(3-甲氧基苯硫基)乙基氨基甲酸苄酯(21)
将化合物20(1.0g)的EtOAc(50mL)溶液冷却至约0℃,加入氯甲酸苄酯(1.1当量)和三乙胺(1.2当量)。将该反应混合物在室温下搅拌4小时,用1N的HCl和饱和NaHCO3(2×10mL)洗涤。通过减压蒸发除去溶剂,获得目标粗产物。用层析法提纯获得白色固体状的21,产率:1.61g。
1H-NMR(300MHz,CDCl3):δ:7.4(s,5H),7.2(m,1H),6.95(d,1H),6.90(s,1H),6.7(d,1H),5.15(s,宽,2H),3.8(s,3H),3.40(t,2H),2.95(t,2H)。
8-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-甲酸苄酯(22)和6-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-甲酸苄酯(23)
将处于苯(5mL)中的化合物21、低聚甲醛(100mg)、对甲苯磺酸(50mg)中的混合物在76℃下搅拌20小时。过滤,然后浓缩,用SiO2层析法提纯,得到比例为50∶1的产物22和23的混合物。总产率:83mg。
1H-NMR(CDCl3):7.80(d,0.5×1H),7.45-7.2(m,6H),7.08(m),6.95(s,0.4×1H),6.80(d),6.68(d),6.70(d,0.6×1H),5.1(s),5.0(s),4.7(s,宽),4.6(s),4.05(s,宽,2H),3.82(s,0.6×3H),3.80(s,0.3×3H),2.8(m,2H)。
实施例12:7-甲基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(26)的合成(方案13)
方案13
2--对甲苯硫基乙胺(24)
将处于CH3CN(30mL)中的4-甲基苯硫酚(1.24g)、2-氯乙胺盐酸盐(2.3g)和Cs2CO3(5.3g)的混合物在50℃下搅拌2天。向反应混合物中加入EtOAc(50mL)和水(30mL)。分离有机相,用水洗涤(2×30mL),减压浓缩获得产物24,产率:1.67g,95%。
1H-NMR(300MHz,CDCl3):δ:7.27(d,2H),7.13(d,2H),3.0(t,2H),2.8(t,2H),2.3(s,3H)。
2-(-对甲苯硫基)乙基氨基甲酸苄酯(25)
向化合物24(1.60g)的EtOAc(100mL)溶液中加入氯甲酸苄酯(2.0g,1.2当量)和Cs2CO3(3.2g,3当量)。将该反应混合物在室温下搅拌5小时。加入三乙胺(2mL)并在搅拌下反应3小时。加入水(30mL)后,分离有机相,用1N的HCl(2×30mL)和饱和NaHCO3(2×30mL)洗涤。通过减压蒸发除去溶剂,获得白色固体产物25,产率:2.4g,79.7%。
1H-NMR(300MHz,CDCl3):δ:7.35(m,5H),7.3(d,2H),7.1(d,2H),5.1(s,宽,2H),3.4(s,broad,2H),3.0(t,2H),2.3(s,3H)。
7-甲基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-甲酸苄酯(26)
将处于甲苯(10mL)中的化合物25(120mg)、低聚甲醛(120mg,过量)和对-甲苯磺酸(40mg)的混合物在70℃下搅拌24小时。将反应混合物过滤,用饱和NaHCO3(2×5mL)洗涤。除去溶剂获得产物26,TLC和NMR表明所述产物是纯的。产率:110mg,89%。
1H-NMR(300MHz,CDCl3):δ:7.40(m,7H),6.95(d,1H),5.1(s,2H),4.5(s,2H),4.0(m,2H),2.8(m,2H),2.1(s,3H)。
实施例13:7-甲硫基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(28)的合成(方案14)
方案14
7-甲硫基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(28)
将处于苯(120mL)中的化合物27(3g)、低聚甲醛(3.5g,过量)、对甲苯磺酸(1.5g)的混合物在80℃下搅拌14小时。将反应混合物过滤,用饱和NaHCO3(3×30mL)洗涤。除去溶剂得白色固体化合物28。产率:2.98g,90%。
1H-NMR(300MHz,CDCl3):δ:7.40(m,6H),7.0(m,2H),5.1(s,2H),4.5(s,2H),4.0(m,2H),2.8(m,2H),2.2(s,3H)。
实施例14:7-三氟甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
氢溴酸盐(31)的合成(方案15)
方案15
7-三氟甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-甲酸苄酯(30)
将处于苯(15mL)中的2-(4-(三氟甲氧基)苯硫基)乙基氨基甲酸苄酯(29)(300mg)、低聚甲醛(300mg)-和对甲苯磺酸(100g)的混合物在80℃下搅拌3天。将反应混合物过滤,用饱和NaHCO3(2×5mL)洗涤。除去溶剂得白色固体30。产物用SiO2层析法提纯(石油醚/EtOAc=3/1)。产率:235mg,76%。
1H-NMR(CDCl3):δ:7.6(d,1H),7.4(m,6H),7.1(s,1H),5.1(s,2H),4.5(s,2H),4.0(m,2H),2.8(m,2H)。
7-三氟甲氧基-2,3,4,5-四氢苯并[f][1,4]硫氮杂
氢溴酸盐(31)
将起始物质30(200mg)用3mL HBr/AcOH溶液处理1小时。向该反应混合物中加入乙醚(20mL)。过滤白色固体得目标化合物的HBr盐。产率:182mg。
1H-NMR(300MHz,CD3OD):δ:7.8(d,1H),7.5(s,1H),7.3(d,1H),4.6(s,2H),3.7(m,2H),3.0(m,2H)。
MS:250(M+1)。
实施例15:1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)-丙-2-烯-1-酮(33)(方案16)
方案16
N-(2-(4-甲氧基苯硫基)乙基)丙烯酰胺(32)
向化合物1(1.83g)的EtOAc(50mL)溶液中加入丙烯酰氯(1.35g,1.5当量)和Na2CO3(2.12g,2.0当量)。将该反应混合物在室温下搅拌过夜并加入三乙胺。搅拌20分钟后,将所述混合物用水(20mL)、1N HCl(2×20mL)和饱和NaHCO3(2×20mL)洗涤。通过减压蒸发除去溶剂,获得白色固体产物32(TLC上的一个斑点),产率:2.2g,92.8%。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),6.3(d,1H),6.1(dd,1H),5.95(宽,1H,NH),5.6(d,1H),3.8(s,3H),3.5(m,2H),3.0(t,2H)。
1-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂-4(5H)-基)-丙-2-烯-1-酮(33)
将处于苯(7.0mL)中的化合物32(280mg)、低聚甲醛(600mg)、对甲苯磺酸(140mg)的混合物在80℃下搅拌16小时。将反应混合物过滤,用饱和NaHCO3溶液(2×3mL)洗涤。通过减压蒸发除去溶剂,获得油状产物33。产率:253mg,86%。
1H-NMR(300MHz,CDCl3):δ:7.4(m,1H),7.1(s,1H),6.8(m,1H),6.5(dd,1H),6.3(m,1H),5.7(dd,1H),4.7(s,2H),4.0(s,宽,2H),3.8(s,3H),2.8(m,2H)。
实施例16:7-甲氧基-1,2,11,11a--四氢苯并[f]吡咯并[2,1-c][1,4]硫氮杂-3(5H)-酮(35)的合成(方案17)
方案17
(R)-5-((4-甲氧基苯硫基)甲基)--吡咯烷-2-酮(34)
向(R)-(5-氧代吡咯烷-2-基)甲基4-甲苯磺酸酯(Helvetica Chimica Acta1990,73,122-32;Tetrahedron:Asymmetry 2007,18,664-670)(1.05g,3.9mmol)的CH3CN(30mL)溶液中加入4-甲氧基苯硫酚(0.55g,3.9mmol)和Cs2CO3(5g,过量)。将该反应混合物搅拌3天。向该溶液中加入EtOAc(50mL)和H2O(30ml)。分离有机相。通过蒸发除去溶剂,获得油状目标化合物(0.91g,98%)。TLC和NMR表明所述产物对下一步反应而言是足够纯净的。
1H-NMR(300MHz,CDCl3):δ:7.4(d,2H),6.8(d,2H),6.19(s,br,1H),3.8(s,3H),3.7(m,1H),3.0(dd,1H),2.8(dd,1H),2.3(m,3H),1.80(m,1H)。
7-甲氧基-1,2,11,11a--四氢苯并[f]吡咯并[2,1-c][1,4]硫氮杂
-3(5H)-酮(35)
将处于苯(60mL)中的5-((4-甲氧基苯硫基)甲基)吡咯烷-2-酮(34)(160mg)、低聚甲醛(2.0g)、对甲苯磺酸(200mg)的混合物在80℃下搅拌18小时。将该反应混合物过滤,用饱和NaHCO3溶液(2×10mL)洗涤。通过减压蒸发除去溶剂,获得油状粗产物34。所述产物用SiO2层析法(CH2Cl2/EtOAc=5/1)提纯。产率:140mg,87.5%。
1H-NMR(300MHz,CDCl3):δ:7.45(d,1H),7.05(s,1H),6.7(d,1H),4.95(d,1H),4.6(d,1H),4.0(m,1H),3.8(s,3H),2.9(d,1H),2.6(d,1H),2.4(m,2H),2.3(m,1H),1.6(m,1H)。
实施例17:化合物402-(7-甲氧基-2,3-二氢苯并[f][1,4]硫氮杂
-4(5H)-基)乙酸(ARM111))的合成(方案18)
方案18
如实施例1所述的那样,将化合物1转化成化合物4或其游离碱4a。为制备化合物40,将化合物4或其游离碱4a、1-溴代乙酸甲酯和吡啶的混合物在DMF中反应适当的时间。向该混合物中加入乙酸乙酯,以及如果需要的话,将所述反应混合物用碱性溶液(如NaHCO3)或水洗涤。油状的产物化合物39(ARM110)可用例如SiO2层析法提纯。然后,将碱(如1NNaOH)加入到处于溶剂(如MeOH)中的化合物39中,将该混合物反应适当的时间。然后除去溶剂(例如在减压下进行),然后将残余物溶解到水性溶液,例如水中。水相可用乙酸乙酯洗涤并酸化(例如用1N的盐酸)至pH值约为4。然后,可除去溶剂(例如在减压下进行),制得粗产物40。可采用醇(如乙醇)除去NaCl,得到纯净的固体40。
本发明不限于本文所描述的具体实施方案的范围。实际上,除了本文所描述的实施方案,通过前文的的描述,处于本发明的宗旨和范围内的各种变型,对本领域技术人员而言也是显而易见的。这些变型也落入了所描述的本发明范围之内。